The Importance of Oxidative Stability In Polyolefins, Part 2
The DSC test can do a reasonably good job of capturing the comparative behavior of materials that use similar antioxidant chemistries.
.jpg;width=70;height=70;mode=crop;format=webp)
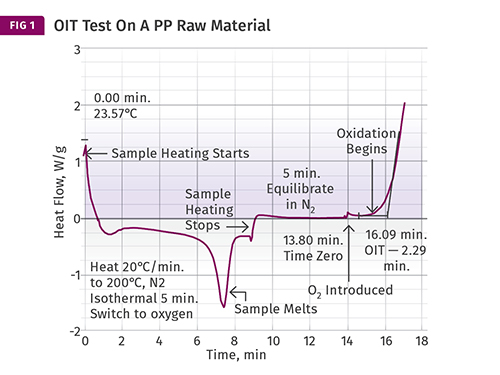
This shows the result of a test for oxidative stability performed on a PP raw material. The test begins in nitrogen at room temperature and the sample is heated to a specific temperature, then oxygen is introduced and the oxidation induction time (OIT) is measured to show how long the antioxidant package protects the polymer from oxidation.
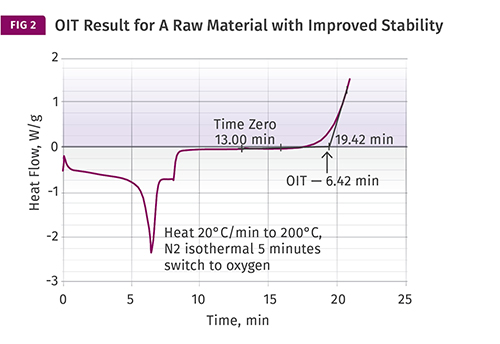
This test result is for a second PP raw material that has somewhat improved oxidative stability, as measured by the oxidation induction time (OIT) compared with the sample in Fig. 1.
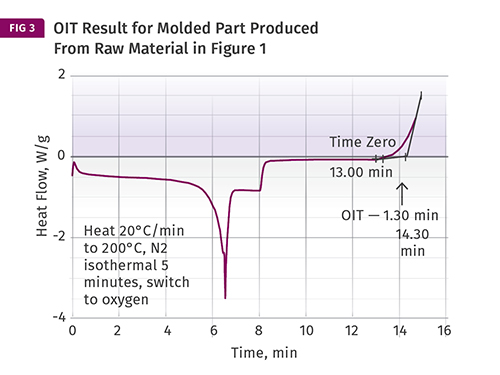
Another use of this DSC test is to compare a raw material to a molded part produced from that material. The oxidation induction time (OIT) for the molded part is approximately 1 min less than that of the raw material in Fig. 1.



In the last column on the topic of oxidative stability in polyolefins, I discussed some of the problems that can arise when oxidation occurs in these materials. I also mentioned that tests were developed to assess the relative oxidative stability of polyolefins using differential scanning calorimetry (DSC). The oxidative stability test was developed in the 1960s and has been around for so long that it is often neglected as a tool for analyzing performance problems.
In principle, the test is relatively simple. A sample of raw material or a piece of a molded part is placed in a sample pan and loaded into the DSC. This instrument measures the absorption or release of heat by a material as a function of temperature or time. DSC is usually employed to evaluate phase changes such as melting and recrystallization and second-order transitions such as the glass transition. It is also used to measure the heat capacity of a material, the energy required to increase the temperature of a substance.
But it can also be used to evaluate any event that involves a change in the heat content of a material. Degradation that involves the evolution of volatiles, such as occurs when a polymer such as acetal (POM) is overheated, is an endothermic (heat-absorbing) process that can be observed using this method. The curing process of a thermosetting polymer such as an epoxy is exothermic (heat releasing) and also registers in a DSC thermogram. Oxidation is also exothermic and in polyolefins it tends to occur relatively quickly, releasing heat at a readily defined point in time.
Figure 1 shows the result of a test for oxidative stability performed on a PP raw material. The test begins in nitrogen at room temperature and the sample is heated to a specific temperature. Virtually any temperature can be used, however the most commonly used standard method uses a temperature of 392 F (200 C). If the material being tested is a PE or a PP, this will place the sample in the molten state.
The melting process for this sample can be observed by the downward deviation from the baseline. Once the melting process is complete, the curve returns to the baseline and the heating process continues until the set temperature of the test is achieved. Once the sample reaches the desired temperature the heating process stops. This is indicated by the step change as noted. The sample is allowed to come to equilibrium with the set temperature for a few minutes, and at that point air or oxygen is introduced into the instrument. At this point the oxygen begins to react with the polymer and its additives. As long as the antioxidant in the polymer is present, it will protect the polymer.
However, once the stabilizer has been consumed by the combined effects of the oxygen and elevated temperature, oxidation occurs and is indicated by the rapid upturn in the baseline. This point is known as the oxidation induction time (OIT) and is calculated from the time of the introduction of the oxygenated atmosphere. In this case the process takes 2.29 min (16.09-13.80 min).
By itself, the result has no intrinsic significance. However, if multiple material samples employing similar stabilizer chemistries are compared, the range of OIT values provides a relative indicator of oxidative stability and can be correlated with the amount of antioxidant that is present in a material. Since quantifying the level of antioxidant in a sample involves relatively sophisticated and time-consuming tests, being able to obtain information regarding oxidative stability this simply is very useful. Fig. 2 shows a test result for a second raw material that has somewhat improved stability compared with the sample in Fig. 1.
Another use of this method is to compare a raw material to a molded part produced from that material. As mentioned previously, melt processing inevitably consumes some of the anti- oxidant in the material. The difference between the OIT of the raw material and the molded part tells us something about the effect that the process has on the stabilizers in the material. Figure 3 shows a test performed on a sample taken from a molded part produced from the raw material tested in Fig. 1. The OIT for the molded part is approximately 1 min less than that of the raw material. If the process conditions are changed, particularly any setting that alters the degree of exposure of the material to elevated temperatures, such as melt temperature, screw rotation speed, or backpressure, this will be reflected in the result we obtain for the molded part.
The effects of post-processing steps such as sterilization or the influence of the application envi- ronment can also be documented by monitoring the change in the OIT as a function of exposure to these conditions. For example, it has been observed that PP exposed to gamma or E-beam sterilization processes may exhibit dramatically reduced OIT values, potentially resulting in a significant loss in toughness. Long-term heat-aging tests designed to accelerate the failure of a product will also have the effect of progressively reducing the OIT of the material.
As has been noted, the test standards tend to employ temperatures that melt the sample. This alters the reaction rate associated with oxidation, causing it to occur much more rapidly than it would when the material is used in the solid state. In addition, melting the sample creates a much greater surface area that is available for interaction with oxygen than would be available in the solid state. This means that the tests are more suited to evaluating process stability than application stability. And even attempting to equate directly what we see in the instrument to what we would observe during processing is not a correlation that can be made readily. While we can send pure oxygen through the DSC, the atmosphere that the part is subjected to is only 21% oxygen.
Balancing this is the fact that in the real world of melt processing, shear is always present to some degree. The mechanical work imparted to the material by the processing equipment adds heat that cannot be replicated in the lab instrument. Yet despite these limitations in simulating a particular environment, the relative differences that are measured with this technique do a reasonably good job of capturing the comparative behavior of materials that utilize similar antioxidant chemistries. Therefore, the approximate threefold advantage that the raw material in Fig. 2 has over the material in Fig. 1 will tend to translate across a wide range of exposure conditions both in the melt and in the solid state.
Next month we will delve into the details of improving the utility of this test by using variations on the standard test methods in order to better understand the oxidation mechanism.
ABOUT THE AUTHOR
Mike Sepe is an independent, global materials and processing consultant whose company, Michael P. Sepe, LLC, is based in Sedona, Ariz. He has more than 35 years of experience in the plastics industry and assists clients with material selection, designing for manufacturability, process optimization, troubleshooting, and failure analysis. Contact: (928) 203-0408 mike@thematerialanalyst.com.
Related Content

The Importance of Melt & Mold Temperature
Molders should realize how significantly process conditions can influence the final properties of the part.
Read More
How Much L/D Do You Really Need?
Just like selecting the extruder size and drive combination, the L/D should be carefully evaluated.
Read More
Tunnel Gates for Mold Designers, Part 1
Of all the gate types, tunnel gates are the most misunderstood. Here’s what you need to know to choose the best design for your application.
Read More
How to Stop Flash
Flashing of a part can occur for several reasons—from variations in the process or material to tooling trouble.
Read MoreRead Next

MATERIALS: The Importance of Oxidative Stability in Polyolefins
Because oxidation is a process that causes materials to deteriorate over time, its effects or potential for those effects are not always apparent when new products are tested.
Read More
Advanced Recycling: Beyond Pyrolysis
Consumer-product brand owners increasingly see advanced chemical recycling as a necessary complement to mechanical recycling if they are to meet ambitious goals for a circular economy in the next decade. Dozens of technology providers are developing new technologies to overcome the limitations of existing pyrolysis methods and to commercialize various alternative approaches to chemical recycling of plastics.
Read More
Troubleshooting Screw and Barrel Wear in Extrusion
Extruder screws and barrels will wear over time. If you are seeing a reduction in specific rate and higher discharge temperatures, wear is the likely culprit.
Read More