
Compounding: Part 2 of 2 Demystifying Devolatilization
Here are some simple tools to help monitor and optimize your devolatilization process.



It is instructive to break apart the controlling variables for devolatilization into machine-design parameters and thermodynamic variables. This helps deconvolute the problems and makes the controlling mechanisms a bit clearer in the mind of the designer and operator.
Machine Parameters
Devolatilization is the process by which solvents or dissolved gases are stripped from a polymer solution or melt. Devices employed for devolatilization are wide ranging in design, from falling-film evaporators to multi-screw extruders, but the guiding principles remain the same. One must create a vapor space above the polymer melt or solution and provide mechanical means for regenerating surface area. In that vapor space, one needs to create conditions that favor movement of volatiles from the melt to the vapor.
Featured Content

While quantification of the devolatilization process is very challenging, the nature of the process lends itself to grouping two sets of parameters: mechanical and thermodynamic. The mechanical variables are equipment design parameters that link the geometry with three key process metrics:
• Surface area and rate of regeneration;
• Residence time;
• Distributive mixing.
Why is surface area so important? Imagine two cups of water of equal volume, taken from the same tap at the same temperature, and placed on a table in a room. Now one of those cups is spilled out over the table while the other is left in the cup. We all know if we return to that room after several hours, that water poured on the table will be largely gone, while the water in the cup will still be largely as we left it. The difference is surface area. A gallon of paint in a can be left open for hours and remain fluid and serviceable. Spread that same paint out over a wall and within a few hours it is dry and ready for pictures and curtains. Again, the difference is surface area.
A volatile species will leave a solution only at a gas/liquid (or melt) interface. The larger the interface, the faster the loss of the volatiles. In the example of water on the table, it need not be above its boiling point and in that case is only at room temperature. It need only have sufficient area and a concentration gradient—the water on the table dries faster on a dry day than on a humid day, the difference being concentration of water vapor in the air.
A volatile species will leave a solution only at a gas/liquid (or melt) interface. The larger the interface, the faster the loss of the volatiles.
So, if surface area is so important, why do we need distributive mixing and surface-area regeneration? The accompanying image shows a profile view of a venting zone in an extruder screw. The channels are partially filled with melt, and it is easy to imagine that the surface of the melt is continually refreshed as the screw rotates. Because of the relatively slow rates of diffusion of the volatiles through the melt, only the molecules very near the surface will escape to the vapor phase. If one were to rely on diffusion of the volatiles through the melt, devolatilization would take weeks or months rather than seconds.
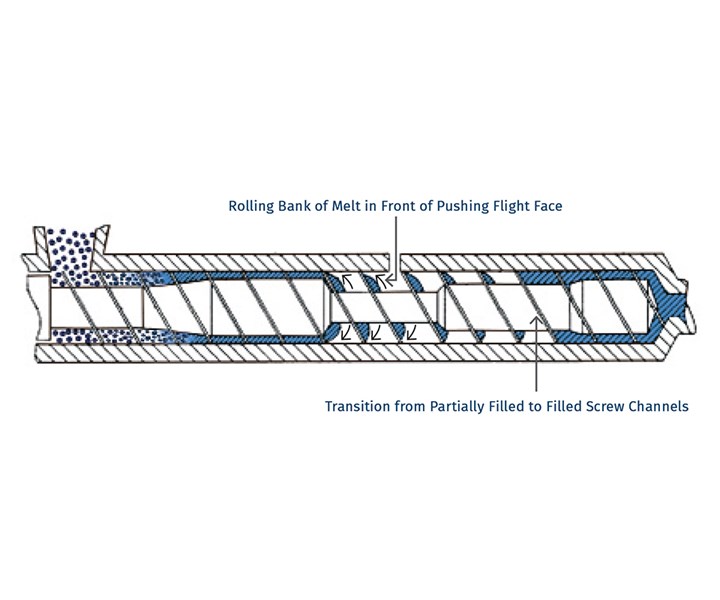 Single-screw devolatilizing extruder with rolling banks of polymer.
Single-screw devolatilizing extruder with rolling banks of polymer.Instead, we help mother nature along by continually bringing fresh material to the surface of the channel via surface-area regeneration, and by ensuring a homogenous mixture in the channel itself by optimizing distributive mixing.
The focus on distributive mixing may seem a bit misplaced at first, but it is important to remember that as the immediate layer at the surface of the melt is depleted of the volatile components, the process shuts down unless that surface is replaced with fresh melt. The diffusion rate of volatiles in the melt relative to the short time in a DV zone is such that a high degree of mechanical mixing needs to occur, coupled with surface-area regeneration to keep the stripping process at its highest rate.
Unfortunately, options for mixing are limited, because one cannot simply put in kneading or other non-conveying elements, as that would risk material backing up into the vent. Instead, we optimize the turnover in the channel, exchange between screws in a twin-screw extruder, and the time in the devolatilization zone. Screws in typical devolatilization sections, whether single- or twin-screw, are partially filled and experience laminar flow, meaning that bringing fresh material to the surface of the rolling bank can be accomplished only through high rates of laminar mixing. Multiple flights, more steeply pitched screws, and interruptions in flights all contribute to maximizing the rate of bringing fresh material to the surface.
What is important to note is that these are all predictable parameters, without having to model bubble nucleation, growth, and rupture. One knows that surface area is important and that the rate of generation of surface area is important. Regardless of bubbles or no bubbles, nothing leaves the melt unless it is at a melt/gas interface.
For example, one can write a simple relationship for generation of surface area per unit time as a function of the number of channels, the geometry, and the screw speed. It is important to remember that the total surface area is the film deposited on the barrel wall, along with the face of the rolling bank of polymer.
These simple relationships show that the rate of surface-area generation increases with the number of channels, screw speed, and decreased degree of channel fill. The pitch of the screw, generally set as a square pitch (one turn of the flight in one diameter of the screw) can be altered to a steeper pitch to improve surface-area generation. This is embedded in the helical length of the zone Z as shown here:
SAc = (Vy) × (nZH) (surface area/unit time in channel)
SAb = (1-f)W × nZ × N (surface area/unit time on barrel surface)
W = channel width
f = fill fraction
Z = helical length of zone
N = screw speed
n = number of channels
H = channel depth
Vy = rolling-bank face velocity
Similar relationships may be developed for residence time in the zone, which contributes to both mixing and surface-area generation. It stands to reason that the longer the polymer is in the vented section, the more volatiles will be removed.
Thermodynamic Variables
The thermodynamic variables (the mere word strikes fear into the hearts of many of us) come down to maximizing the driving force for volatiles to leave the melt or solution and enter the vapor space above the melt.
While thermal energy is one of the primary drivers for devolatilization, too high a temperature for too long a time will harm the polymer in other ways leading to oxidative degradation, etc.
The controlling thermodynamic mechanisms change as the concentration of the volatile component changes. Very high concentrations of solvent or monomer behave much like a boiling process. Here the controlling mechanism is driving heat into the melt or solution and allowing a rapid decompression. The elements of the screw design have little impact on this, other than to knock down the foam and prevent flying polymer in the vents.
Very high concentrations of solvent or monomer behave much like a boiling process.
As the concentration of volatiles diminishes, the mechanism moves to a diffusion-controlled mechanism. There is lots of discussion about superheat in the literature and while sufficient energy must be applied to overcome the heat of vaporization etc., this is seldom the limiting step other than in the initial venting operation for a low-solids feed. Instead, the controlling mechanisms are the aforementioned surface-area generation coupled with the partial pressure of the volatilizing species.
Partial pressure is a critical variable in predicting the separation of a gas from a polymer melt. Gases will want to equalize their pressure between two phases that are open to one another. A gas will move from an area where its partial pressure is higher to an area where its partial pressure is lower. In addition, the greater the partial-pressure difference between the two areas, the more rapid is the movement of gases.
The movement from the melt to the vapor space above the melt is dictated by the solubility of the gas in the melt and the partial pressure of that component above the melt. This is the fundamental relationship spelled out in Henry’s law, which one may remember from high school or college physics. Henry’s law states that the concentration of gas in a liquid is directly proportional to the solubility and partial pressure of that gas. The greater the partial pressure of the gas, the greater the number of gas molecules that will dissolve in the liquid. The concentration of a gas in a liquid is also dependent on the solubility of the gas in the liquid.
Leaking systems, inadequate vacuum levels, or the wrong types of vacuum pumps can all dramatically reduce the performance of a vent.
The Flory-Huggins equation shows the relationship between how much of a volatile component wants to stay in the melt and the vapor pressure above the melt. While neither the Flory-Huggins equation nor Henry’s law are directly applicable to a DV vent, qualitatively they can teach a lot:
log(P1/P01) = log(1-V2) + V2 + XV22
P1 = vapor pressure above the melt
P01 = vapor pressure of pure substance at the temperature under consideration
V2 = volume of polymer in binary mixture
X = Chi or Flory-Huggins parameter
From the thermodynamics side, one simply wants to eliminate as much of the volatile component from the vapor space above the melt as possible. This translates to:
• As high a vacuum level as conditions will permit;
• Use of inert sweep gases (nitrogen) to further reduce the concentration of the stripping species above the melt;
• Put energy into the melt to compensate for cooling created by the stripping action.
One can condense all the math into some simple rules of thumb:
• Maximize surface area and regeneration of surface area.
• Ensure the highest degree of distributive mixing in the partially filled channels.
• Reduce the concentration of the volatile species above the melt by employing high vacuum levels and possible sweep gases.
• Stage the stripping (multiple vents) to reflect the changes in controlling mechanisms.
• Keep the viscosity low to minimize degradation.
• Keep oxygen out of the system to minimize degradation.
Practical Matters Matter
There are a variety of practical aspects for good devolatilization performance, including having the proper number of stages, preventing polymer buildup in vents, flying polymer, cooling of the melt as a result of too rapid stripping action. There are several continually recurring themes for poor devolatilization. These include:
• Flying polymer: When the rate of volatile removal is high, devolatilization is more of a boiling process, generating significant amounts of foam and bubbles. As the solution decompresses, gases escape, bubbles are formed and rupture, and there is significant loss of temperature due to the cooling effects of the heat of vaporization of the volatilizing species. This often results in the foam solidifying and coming off the screws and being entrained into the venting system with all the expected undesirable consequences. Allow sufficient screw length after the upstream seal prior to the vent opening to allow the screws to break down the foam and reheat the melt.
• Polymer backing up into the vent: This is a result of too long a filled length (or too short a pumping section) downstream of the vent prior to the downstream seal. Increase the screw speed, improve the pumping efficiency of this section (shallower screw channel), or simply increase the length of screw between the vent opening and the downstream seal. Decrease the pressure drop over the downstream seal so there is less of a ΔP to pump against.
• Thermal regeneration: Stripping large masses of volatiles will significantly cool the melt, perhaps not to the point of solidification, but certainly to the point where further volatilization is impaired. The pumping section and the melt seal downstream of a venting section can be designed to impart more viscous heating to the melt so that the subsequent devolatilization zone operates more efficiently.
• Poor vacuum: Leaking systems, inadequate vacuum levels, or the wrong types of vacuum pumps can all dramatically reduce the performance of a vent. We focus on the extruder while often ignoring the ancillary components that are equally important to this stripping operation.
• Too much in one step: There are practical limits to the reduction in volatiles that can be accomplished in one zone. The beauty of an extruder, especially a twin-screw, is that you can stage the devolatilization vents and optimize the performance of each one. It is not realistic to expect to remove much more than an order of magnitude in concentration per vent—and not necessary, either. Stage the stripping, because it allows the greatest process flexibility and greatest turn-up and turn-down ratios for the extruder.
• Creating new volatiles: Understand the degradation mechanisms for your polymer. Be careful not to subject the melt to conditions that will cause it to unzip, as it is a battle you will not win. Avoid oxygen, and design the stripping stages so that the last vent before discharge is operating at close to capacity. This maintains a lower viscosity throughout the machine and minimizes degradation in the extruder.
What Devolatilization Does Not Do
We have discussed how DV works and how to improve DV performance, but it is worth noting that there are limits to what DV can remove.
What devolatilization will not do is remove non-volatile contaminants from a polymer melt. Larger molecules, dimers, and trimers that have a high boiling point and/or low vapor pressure will not be extracted by devolatilization and must be removed via extraction or reactive processes. The higher the boiling point of a contaminant, the higher the melt temperature and rates of surface-area regeneration, and the lower the partial pressure, must be to strip those materials.
ABOUT THE AUTHOR: Rob Jerman has developed twin-screw extrusion systems for devolatilization, mixing and compounding, and reactive systems for over 30 years and has significant experience in product and process development in fluoropolymers. He has his own consulting firm, Extrusion Technology and Innovations, part of Extrusion Edge. Contact robertjerman@extrusionti.com
















Related Content

Fundamentals of Polyethylene – Part 6: PE Performance
Don’t assume you know everything there is to know about PE because it’s been around so long. Here is yet another example of how the performance of PE is influenced by molecular weight and density.
Read More
Injection Molding: Focus on these Seven Areas to Set a Preventive Maintenance Schedule
Performing fundamental maintenance inspections frequently assures press longevity and process stability. Here’s a checklist to help you stay on top of seven key systems.
Read More
How to Select the Right Cooling Stack for Sheet
First, remember there is no universal cooling-roll stack. And be sure to take into account the specific heat of the polymer you are processing.
Read More
The Fundamentals of Polyethylene – Part 2: Density and Molecular Weight
PE properties can be adjusted either by changing the molecular weight or by altering the density. While this increases the possible combinations of properties, it also requires that the specification for the material be precise.
Read MoreRead Next

Solve Venting Problems on Twin-Screw Compounding Extruders
Compounds must be free of voids and volatiles or else the material will cause subsequent processing problems. Here’s how to ensure your venting is effective.
Read More
Compounding: Part 1 of 2: Demystifying Devolatilization
Very little has been written about this critical topic, and most of what has been published is theoretical. Here’s some practical guidance to help you better understand the process and improve your operation.
Read More
Extrusion Know-How: The Whys & Hows (& Ifs) of Vented Extruders
Vented or two-stage screws only make sense when volatiles must be removed in order to make a satisfactory product.
Read More